
妙啊!用扩散模型生成蛋白质结构,结果不输天然蛋白质|来自斯坦福&微软
网友:未来5年用文本提示生成新抗体/新酶也不是梦吧
丰色 发自 凹非寺
量子位 | 公众号 QbitAI
没想到,图像生成领域的大明星——
扩散模型,这么快就被用来做蛋白质结构生成了!
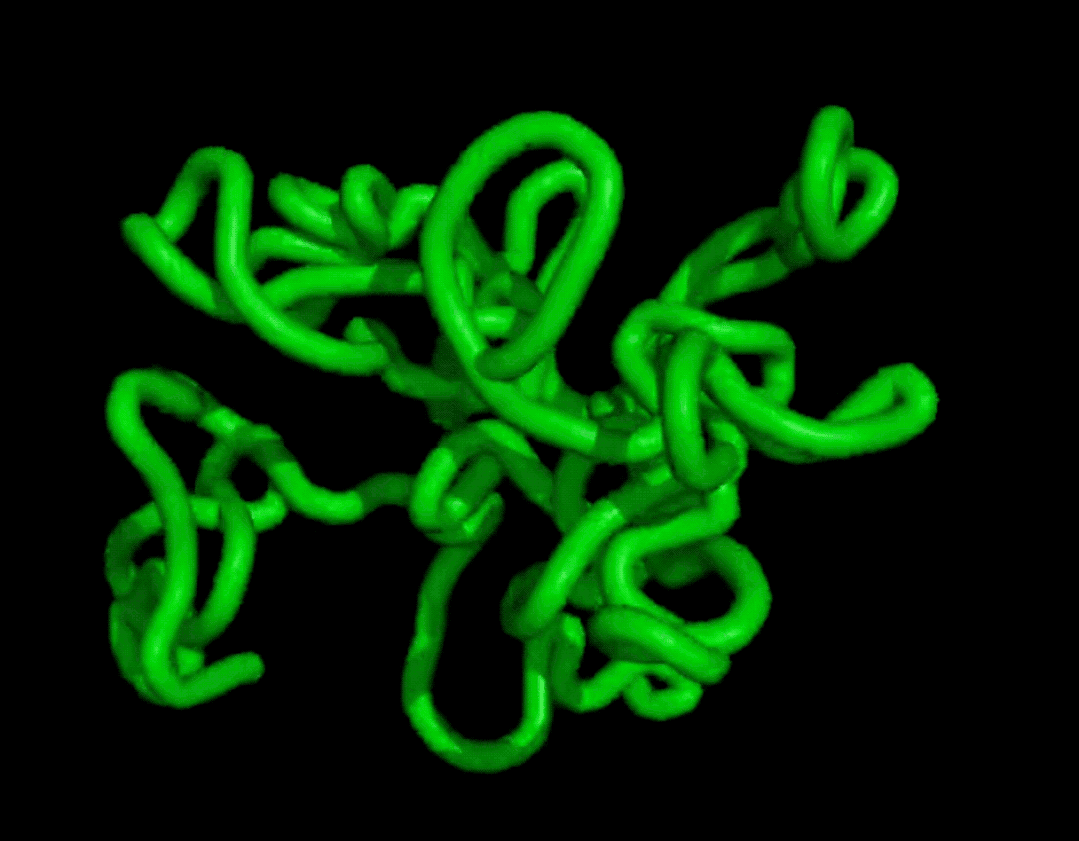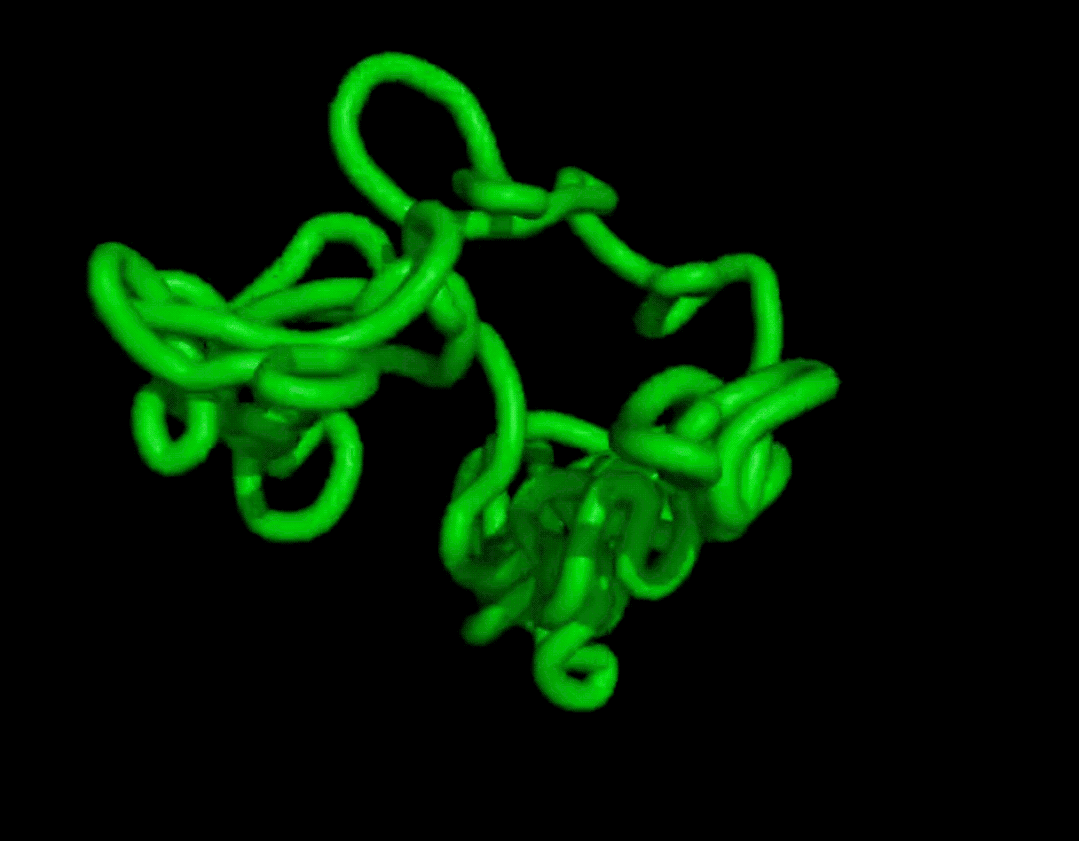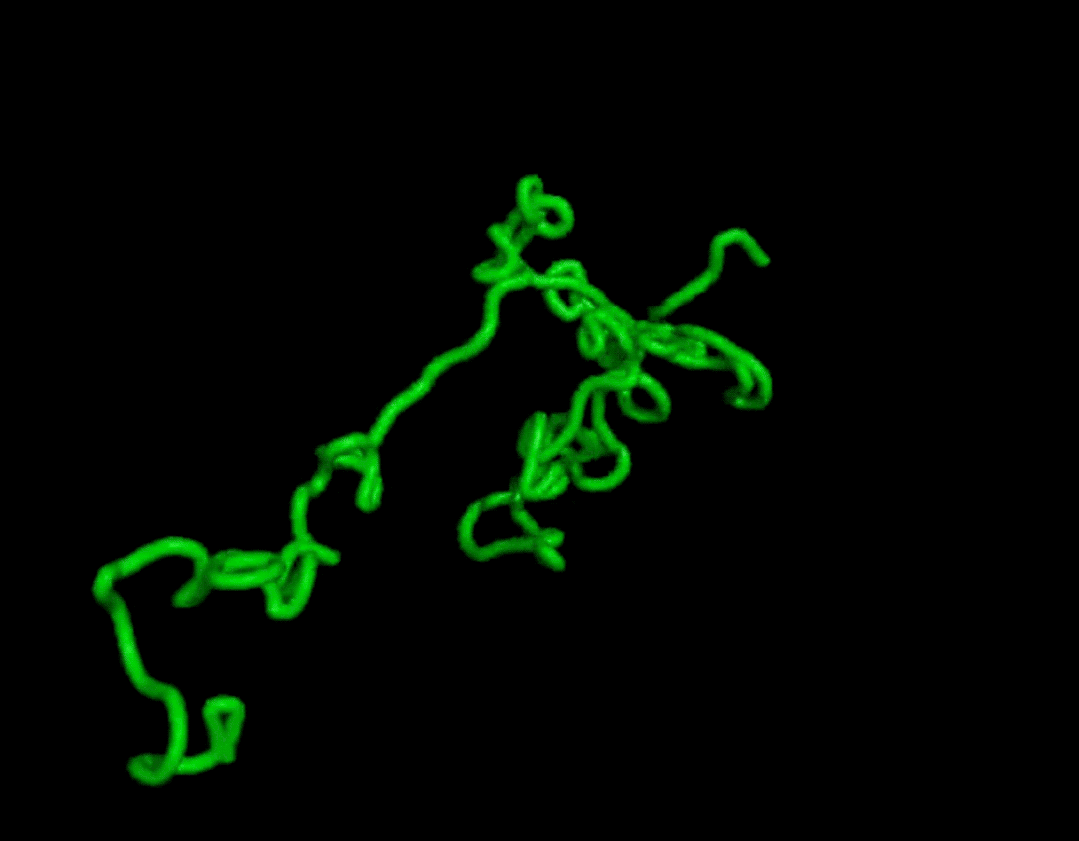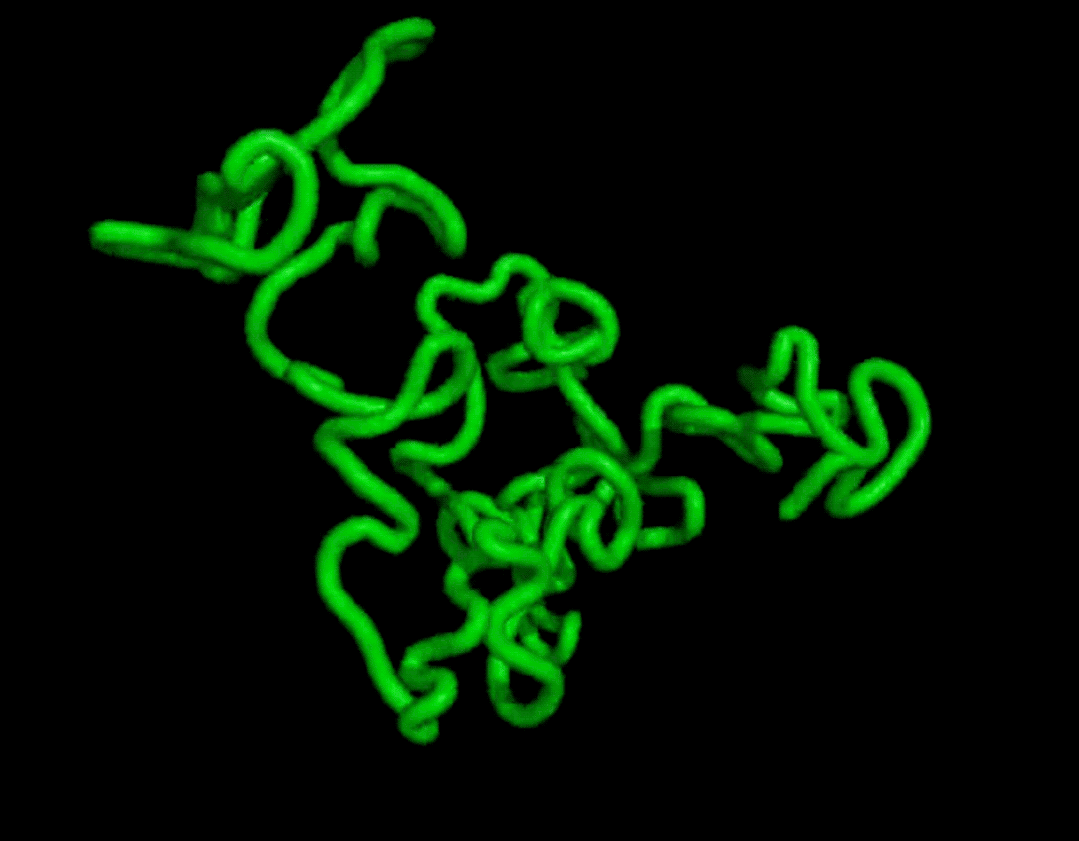
而且结果在复杂度和结构上都和天然蛋白质有的一拼。

消息一出,不少人都称赞这个组合简直非常妙。

还有人表示:我早就猜到了,生成模型能做的真的不仅是图像和视频。
所以,AlphaFold这是可能有新的挑战者了?

具体是怎么回事?
来看看斯坦福大学和微软的这项最新研究成果到底怎么说。

扩散模型vs蛋白质结构生成
说起研究的初衷,作者表示:
尽管蛋白质结构预测已经取得了非常好的成绩,但要从神经网络中直接生成多结构多样又新颖的蛋白质结构仍然很困难。
他们想到用基于扩散的生成模型来挑战这一任务,并通过镜像蛋白质自然折叠过程来设计蛋白质主链结构。
具体来说,就是将蛋白质主链结构看成一系列连续的角度,这些角度会捕捉组成氨基酸残基的相对方向。
进而通过从随机、未折叠状态到稳定折叠结构的去噪就可以生成新结构。
作者表示,这一设计不仅可以反映蛋白质如何在生物学上扭曲成能量上有利的结构(how proteins biologically twist into energetically favorable conformations),这种表示的固有位移和旋转不变性也可以极大地减轻模型对复杂等变网络的需要。
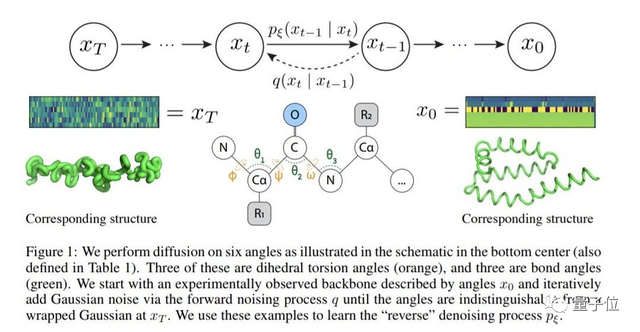
在实现上,作者仅用一个简单的transformer作为backbone就训练出了一个去噪扩散概率模型。
最终证明它可以无条件地生成高度真实的蛋白质结构,其复杂性和结构模式类似于天然蛋白质的结构模式。
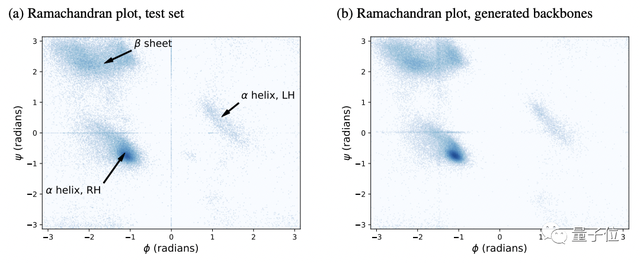
如下图Ramachandran plot(拉氏图,专门用于检测蛋白质构象是否合理)所示,左右分别为测试集和生成的蛋白质主干的(φ,ψ)二面角。
可以看到,三个主要结构元素、以及一些不太常见的角度组合都在他们用扩散模型生成的主干中得到了呈现。
目前,基于以上成果,作者已公开发布了首个用于蛋白质结构扩散的开源代码库和训练模型,详情可戳文末链接。
不过,作为一个初步探索,他们也指明这项成果还存在几个局限性,比如:
1、与通常有几百个残基的天然蛋白质相比,模型生成的结构仍然相对较短(最多128个残基);
2、由于没有处理多链复合物或配体相互作用,模型无法捕获蛋白质的动态性质,只能生成静态结构;
3、将蛋白质表述为一系列角度的框架设计会造成一些累积误差,最终显著改变生成的整体结构。
最后一个问题则可以对未来工作提供思路,比如试着用几何信息架构中使用的方法来解决相关问题。
最后,除了再次刷新我们对扩散模型的认知,一位毕业于ETH的研究人员看完这项成果后还大胆预测道:
未来5年内,可能渐渐没有人会对全新的蛋白质序列或折叠感到兴奋了。
因为潜在的新酶和治疗性抗体将通过文本提示生成。

对于这项成果,你有什么想说的?
论文地址:
https://arxiv.org/abs/2209.15611
代码:
https://github.com/microsoft/foldingdiff
- 北大开源最强aiXcoder-7B代码大模型!聚焦真实开发场景,专为企业私有部署设计2024-04-09
- 刚刚,图灵奖揭晓!史上首位数学和计算机最高奖“双料王”出现了2024-04-10
- 8.3K Stars!《多模态大语言模型综述》重大升级2024-04-10
- 谷歌最强大模型免费开放了!长音频理解功能独一份,100万上下文敞开用2024-04-10
相关阅读











